



推荐产品





公司新闻/正文
方法汇总 | 单细胞转录组联合公共数据库数据挖掘思路大汇总
人阅读 发布时间:2023-01-31 17:04
近年来,随着单细胞测序技术的发展, 单细胞转录组在研究中应用越来越多,而过去多年来,公共数据库中数据积累也非常可观,如批量转录组数据等,因此在科学研究中,在已有单细胞转录组数据的情况下,如何有效联合公共数据库中的数据来阐述相关科学问题是一个值得探讨的话题。
我们今天将对单细胞转录组联合公共数据库进行数据挖掘的常见思路进行简单的汇总,供大家参考。
研究思路一
利用scRNA-Seq数据指导批量RNA-seq数据的挖掘
1.scRNA-Seq揭示了炎症性癌症相关的成纤维细胞在膀胱尿路上皮癌中的作用

在2020年发表的一篇研究膀胱尿路上皮癌的文章中,作者对8个膀胱癌(BC)肿瘤样本与3 个癌旁样本(para tumor samples)进行scRNA-Seq,并在BC微环境中鉴定了19种不同的细胞类型,表明高肿瘤内异质性。文章发现肿瘤细胞中MHC-II分子下调,表明肿瘤细胞的免疫原性下调可能有助于形成免疫抑制微环境。文章还发现单核细胞在肿瘤区域进行M2极化并分化。
此外,LAMP3+DC细胞亚群可能能够募集调节T细胞,参与肿瘤微环境免疫抑制的形成。通过结合公共数据集中的3000多个BC批量RNA-seq数据进行相关性分析,文章确定了炎症性癌症相关的成纤维细胞(ICAFs)在肿瘤进展中的作用,ICAFs与预后不良显著相关。此外,文章还根据ICAFs建立了一个调控网络。这些结果可以帮助阐明ICAF的原始机制,为肿瘤免疫学提供了深刻的见解,并为未来的药物发现提供了重要的资源。
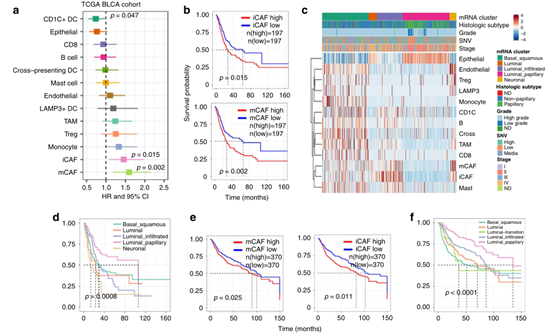
文章以scRNA-Seq为参考数据集,使用CIBERSORTx对3000多膀胱癌的批量测序数据数据反卷积,预测获取了每个TCGA样本的细胞类型丰度。以细胞类型丰度的50%占比为标准,将样本分为某种细胞类型高表达或低表达。使用多因素COX回归分析,计算TCGA 数据中相对细胞丰度与患者生存的相关性,筛选感兴趣的细胞类型进行Kaplan-Meier法生存分析。结合TCGA分子亚型划分,识别细胞丰度改变,最后文章以同样的方法在microarray数据中也进行了验证。
2.基于肿瘤scRNA-Seq与批量RNA-seq的贝叶斯整合分析-BayesPrism软件对细胞类型和基因表达进行反卷积

以往使用scRNA-Seq数据反卷积bulk数据的方法主要为CIBERSORTx,然而CIBERSORTx目前仅有在线工具,需要教育邮箱注册上传数据,且个人分析空间有限,很受数据量大小与国内网速影响。于2022年发表在《Nature Cancer》上的BayesPrism软件,是一种本地化工具,可将scRNA-Seq数据作为先验信息来预测来批量RNA-Seq中细胞类型的组成和基因表达。
在该文章中,作者对胶质母细胞瘤(GBM)、头颈部鳞状细胞癌(HNSCC)和皮肤黑色素瘤(SKCM)进行了分析,以将细胞类型组成与不同肿瘤类型的临床结果相关联,探索恶性和非恶性细胞状态的空间异质性。
研究思路二
基于批量RNA-seq数据辅助scRNA-seq数据细胞类型鉴定
1.通过整合批量RNA-seq和scRNA-seq数据来识别与表型相关的亚群

单细胞RNA测序(scRNA-seq)可以区分异质组织中的细胞类型、状态和谱系。然而,目前的单细胞数据不能直接将细胞群体与特定的表型联系起来。比如,对于单细胞中的恶性细胞可用infercnv识别,但是在癌症和非癌症疾病中,除了肿瘤与正常之外,还有各种各样的外部表型,如治疗抵抗、疾病分期、生存结果和年龄,这些都可以通过批量RNA-seq数据广泛获得,但是难以直接通过单细胞数据本身推测。
于2022年发表在Nature Biotechnology上的一篇算法文章中,作者针对以上问题,开发了Scissor算法。Scissor利用批量RNA-seq数据中的表型信息来识别与表型最高度相关的细胞亚群。Scissor不需要对单细胞数据进行任何无监督的聚类,避免了对细胞簇数或聚类分辨率的主观决定的影响。
Scissor提供了一个灵活的框架来整合批量RNA-seq数据中的各种外部表型,以指导单细胞数据分析,使临床和生物意义相关的细胞亚群能够在没有假设的情况下进行识别:
(1)识别肿瘤细胞。以TCGA肺腺癌为参考数据集,生存时间为表型信息,Scissor在单细胞数据中识别一个有侵袭性肿瘤细胞的亚群,该亚群与较差的预后相关,并表征为低氧相关基因的过表达,据此文章推测:低氧活性推动肺腺癌的进展;
(2)识别免疫治疗相关的T细胞亚群。以黑色素瘤为参考数据集,免疫治疗效果为表型信息,Scissor在单细胞数据中识别了105个T细胞为Scissor+细胞,它们与良好的免疫治疗反应相关;
(3)识别Alzheimer's相关细胞亚群。以7个AD患者,7个正常对照为参考数据集,Scissor在7432少突胶质细胞中识别206个AD阳性细胞,104个normal阳性细胞。在1078个少突胶质前体细胞中识别20个AD阳性,201个normal阳性细胞;在2171个星形胶质细胞中识别179个AD阳性与14个nomal阳性。
2.通过整合批量RNA-seq,来识别与胃腺癌恶性肿瘤细胞

于2021年发表在Gut上的一篇研究胃腺癌的文章中,作者对9个肿瘤和3个非肿瘤样本的27677个细胞进行了scRNA-seq测序,并使用大规模组织学分析和bulk转录数据集对分析结果进行验证。在对单细胞数据进行细胞类型注释后,文章选取上皮细胞(4776个)进行恶性与非恶性细胞区分。文章使用常规的CNV进行区分,4/4776的假定非恶性细胞显示异常的CNV信号,然而只有25.0%的假定恶性细胞表现出高水平的CNV。这一结果得到TCGA的验证,即:大部分原发性胃腺癌样本经全外显子测序发现CNV信号较低,使用CNV难以区分上皮细胞的恶性与非恶性。
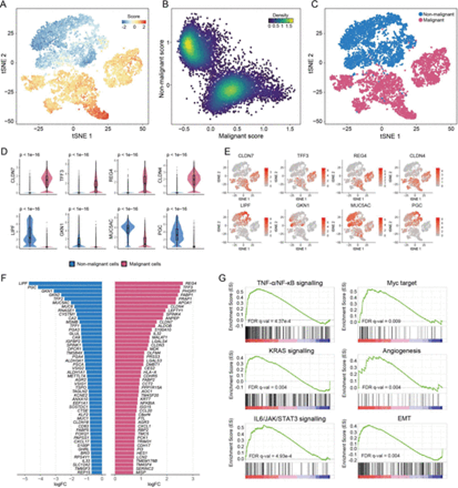
因此,文章通过使用TCGA 肿瘤与癌旁的批量RNA-seq数据集,进行limma差异基因筛选,获取Top50肿瘤特异性高表达基因与Top50正常组织特异性高表达基因对单细胞上皮细胞类型打分(addmodulescore),并对这两列打分结果使用K-means聚为初始潜在恶性与初始潜在非恶性细胞。由于TCGA bulk的初始识别因含有非上皮细胞而产生偏差,文章又重计算两类上皮之间的差异基因,依然用Top50恶性细胞特异性高表达基因与Top50非恶性细胞特异性高表达基因对所有上皮细胞打分,根据此打分重聚类。该重聚类结果与上一次聚类结果做比较(比如同一聚类簇中barcodes重叠占比),若聚类结果相差较大则重复打分聚类步骤,直到聚类结果趋于稳定(比如多次聚类,同类型簇中barcodes重叠率达90%以上)。使用该方法,文章鉴定出5635个恶性上皮细胞和4776个非恶性上皮细胞。后续对此聚类着色作图,差异基因富集,都验证了该方法结果的准确性。
研究思路三
基于药物靶标数据对scRNA-Seq数据进行注释
scRNA-seq揭示了新生儿室管膜瘤的细胞层次结构和异常发育轨迹

室管膜瘤是一种中枢神经系统肿瘤的异质性实体瘤。于2022年发表在Cancer Cell上的一篇研究室管膜瘤的文章中,作者应用scRNA-Seq来分析不同分子类群和解剖位置之间的室管膜瘤,以调查它们在肿瘤内的异质性和发展源头。室管膜瘤由起源于未分化群体的细胞层次结构组成,这些细胞群体经历了三种向神经元-神经胶质命运谱系分化受损的过程。预后良好的室管膜瘤细胞亚型主要含有高度分化细胞,而侵袭性细胞亚型则存在更多的未分化细胞群。文章描述的转录特征与患者生存相关,并定义了靶向治疗方法的分子依赖性。
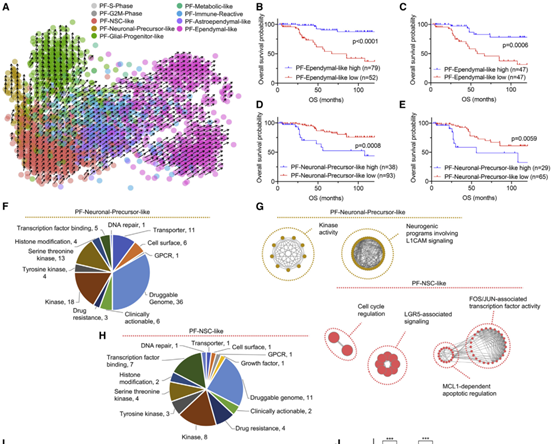
文章进一步使用infercnv筛选恶性细胞,对恶性细胞进行非负矩阵分解(NMF),并以NMF所得的9个元模块对细胞打分,以最高分注释细胞类型。使用分别有高低分化水平的PF-Ependymal-like和PF-Neuronal-Precursor-like 结合TCGA数据预后,发现高分化水平有较好预后,而低分化水平则相反。选取两个低分化水平的模块:PF-Neuronal-Precursor-like和FC-NSC-like。对PF-Neuronal-Precursor-like与药物基因互作数据库(DGIdb)进行整合,使用数据库信息注释模块中基因,并显示了”Druggable genome“ 和 “Clinically actionable”相关基因。对模块与数据库交集基因进行富集与通路分析,明确被注释基因的生物学功能。这些药物干预的靶基因可作为后续用药的指导与预测。
研究思路四
结合疾病相关基因集探寻病理
单细胞分辨率下人类肠道发育的时空分析

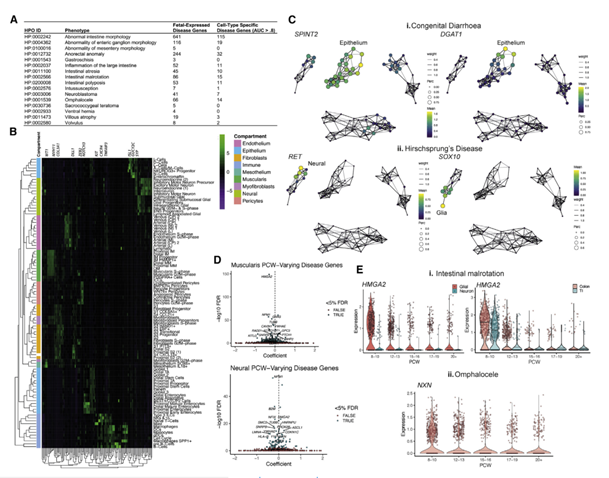
先天性肠道疾病的发病机制仍不明确,因为基础遗传缺陷的疾病较为稀少,并且有许多发生在胎儿尚在子宫中的早期。为了揭示可能导致先天性肠道疾病发育时间特异性的转录缺陷,文章将自身单细胞转录组数据与人类表型本体论数据库(Human Phenotype Ontology ,HPO)中用遗传表型注释的围产期肠道疾病的列表进行了关联,探寻先天性肠道疾病的发病机制。文章筛选749个肠道疾病相关基因,其中718个在文章数据中表达。使用AUCELL计算每一个基因集在数据中的评分,每一个细胞都将获得对应打分的基因集与其对应的AUC值。对每一个基因集在cluster(可以是组织类型注释,细胞类型注释,时间注释等等)下做AUCell.AUC>0.8则该基因为细胞类型特异性的。该基因缺陷则可导致相应疾病。对筛选出的特异性基因基于发育时间点做差异表达,则可筛选出发育时间特异性基因。
研究思路五
利用公共数据集验证细胞类型存在
内皮细胞的onco-fetal重编程驱动肝细胞癌中的免疫抑制巨噬细胞

长久以来,样本量不足一直是个困扰研究者的问题,在此基础上的发现,也会因此而遭受质疑。除了联合bulk数据与临床验证外,使用已发表的单细胞数据验证也是很好的策略。
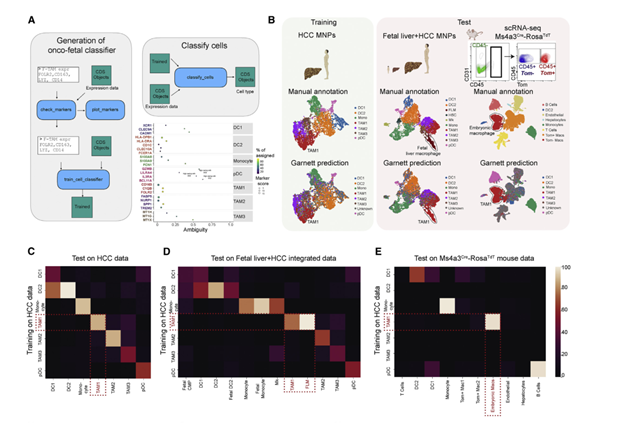
文章发现了内皮重编程驱动的肿瘤相关性巨噬细胞(TAM)类群,但是TAM有可能是单核细胞来源的,也有可能是胚胎驻留的,且胚胎组织驻留巨噬细胞和单核细胞来源巨噬缺乏可靠marker进行区分。因此文章作者团队培育Ms4a3Cre-RosaTdT小鼠(RNA-Seq测序),用以区分胚胎驻留(Tomato– )和单核细胞来源的巨噬细胞(Tomato +)。以肿瘤中单核吞噬细胞(mononuclear phagocytes,MNPs)为训练集,以胎肝,转基因小鼠肝为测试集。结果表明,转基因小鼠embryonically macro( Tomato–)与肿瘤TAM1有高度关联;胎肝巨噬细胞(FLM)与肿瘤TAM1有高度关联。这证明TAMs中的确有细胞类群经历了胚胎式重编程。
总结
今天我们就单细胞转录组联合公共数据库进行数据挖掘的常见分析思路进行了汇总,现在我们来做一下简单回顾,主要包含5个方面:
(1)基于scRNA-Seq数据,指导批量RNA-seq数据的挖掘,以获得批量RNA-seq数据中包含的更多的潜在信息;
(2)基于批量RNA-seq数据,结合临床信息,辅助对scRNA-Seq数据进行细胞类型注释,使得结果更为可信,更具临床价值;
(3)结合药物靶标数据库,有利于指导、预测后期试验用药与药效,推进“老药新用”等测试方案;
(4)对于发病机制仍不明确的疾病,结合疾病相关基因数据库可探寻个体发育、疾病发展过程中基因失调机制,从而揭示转录缺陷;
(5)利用公共数据库中单细胞转录组数据,对通过现有scRNA-Seq数据鉴定的细胞类型进行验证。
1.Chen Z, Zhou L, Liu L, et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun. 2020 Oct 8;11(1):5077.
2.Chu T, Wang Z, Pe'er D, et al. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat Cancer. 2022 Apr;3(4):505-517.
3.Sun D, Guan X, Moran AE, et al. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat Biotechnol. 2022 Apr;40(4):527-538.
4.Zhang M, Hu S, Min M, et al Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut. 2021 Mar;70(3):464-475.
5.Gojo J, Englinger B, Jiang L, et alet al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell. 2020 Jul 13;38(1):44-59.e9.
6.Fawkner-Corbett D, Antanaviciute A, Parikh K, et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell. 2021 Feb 4;184(3):810-826.e23.
7.Sharma A, Seow JJW, Dutertre CA, et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell. 2020 Oct 15;183(2):377-394.e21.
详细技术请访问欧易生物官网
⬇
百度搜索欧易生物(oebiotech)
⬇
了解更多多组学技术
(单细胞转录组测序)
长按扫码
咨询欧易生物技术工程师
询价列表

暂时没有已询价产品
